B. REKAIBI¹ ; S. HECHCHAD² ; N. BOUTRID¹
¹ Service de pédiatrie EHS El-Eulma ; ² Pôle pédiatrique, CHU de Sétif
Introduction :
Les cardiopathies congénitales (CC) revêtent des présentations variées chez les enfants aux traits syndromiques, justifiant un dépistage systématique. Au-delà des associations classiques comme la T21, d’autres syndromes rares révèlent des CC spécifiques.
Objectif :
Attirer l’attention sur les syndromes les moins fréquents pour optimiser le diagnostic précoce.
Observations :
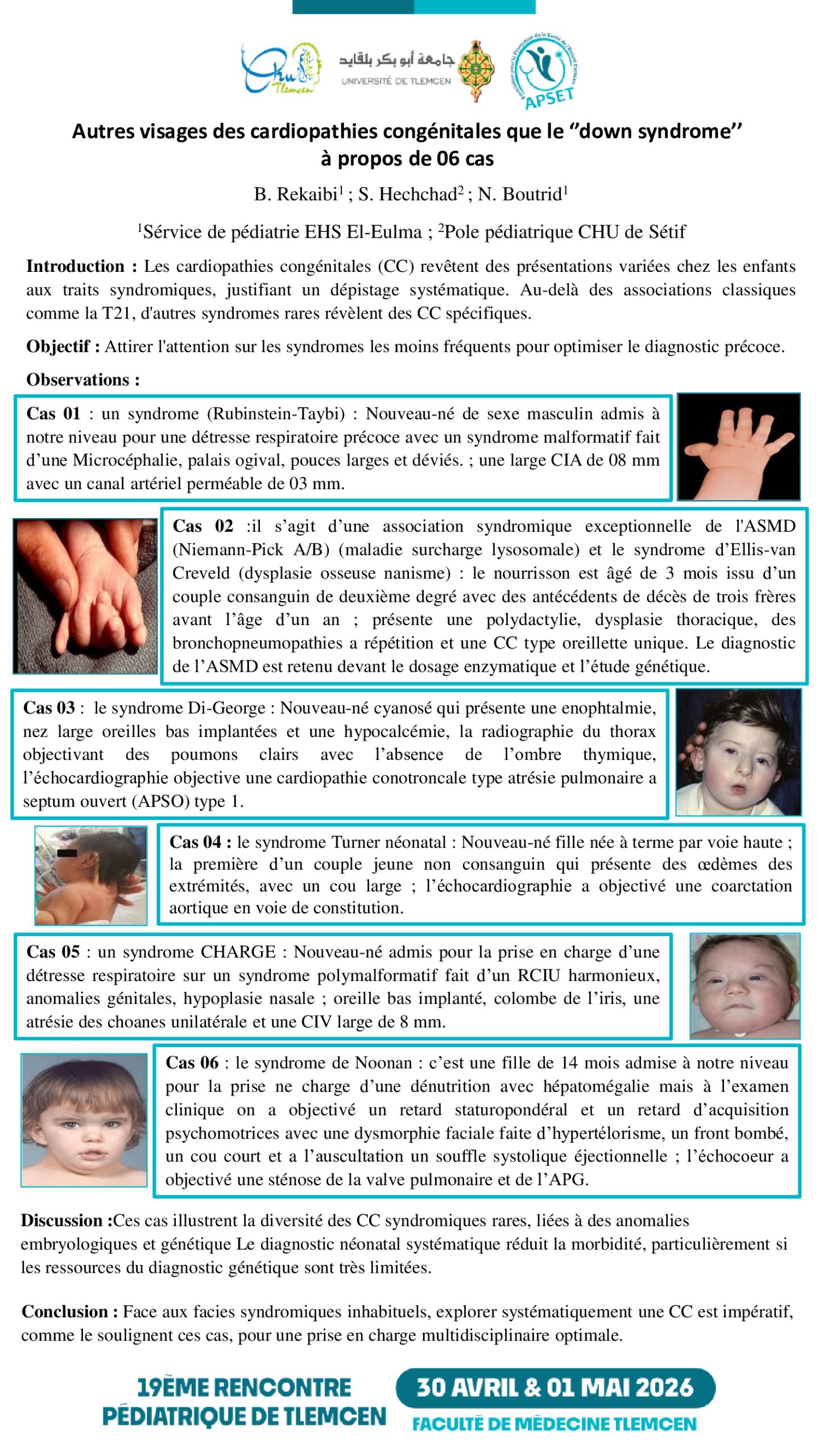
Cas 01 : un syndrome (Rubinstein-Taybi) : nouveau-né de sexe masculin admis à notre niveau pour une détresse respiratoire précoce avec un syndrome malformatif fait d’une microcéphalie, palais ogival, pouces larges et déviés ; une large CIA de 08 mm avec un canal artériel perméable de 03 mm.
Cas 02 : il s’agit d’une association syndromique exceptionnelle de l’ASMD (Niemann-Pick A/B) (maladie de surcharge lysosomale) et le syndrome d’Ellis-van Creveld (dysplasie osseuse nanisme) : le nourrisson est âgé de 3 mois, issu d’un couple consanguin de deuxième degré avec des antécédents de décès de trois frères avant l’âge d’un an ; présente une polydactylie, dysplasie thoracique, des bronchopneumopathies à répétition et une CC type oreillette unique. Le diagnostic de l’ASMD est retenu devant le dosage enzymatique et l’étude génétique.
Cas 03 : le syndrome Di-George : nouveau-né cyanosé qui présente une énophtalmie, nez large, oreilles bas implantées et une hypocalcémie ; la radiographie du thorax objectivant des poumons clairs avec l’absence de l’ombre thymique ; l’échocardiographie objective une cardiopathie conotroncale type atrésie pulmonaire à septum ouvert (APSO) type 1.
Cas 04 : le syndrome Turner néonatal : nouveau-né fille née à terme par voie haute ; la première d’un couple jeune non consanguin qui présente des œdèmes des extrémités, avec un cou large ; l’échocardiographie a objectivé une coarctation aortique en voie de constitution.
Cas 05 : un syndrome CHARGE : nouveau-né admis pour la prise en charge d’une détresse respiratoire sur un syndrome polymalformatif fait d’un RCIU harmonieux, anomalies génitales, hypoplasie nasale ; oreille bas implantée, colobome de l’iris, une atrésie des choanes unilatérale et une CIV large de 8 mm.
Cas 06 : le syndrome de Noonan : c’est une fille de 14 mois admise à notre niveau pour la prise en charge d’une dénutrition avec hépatomégalie, mais à l’examen clinique on a objectivé un retard staturo-pondéral et un retard d’acquisition psychomotrice avec une dysmorphie faciale faite d’hypertélorisme, un front bombé, un cou court et à l’auscultation un souffle systolique éjectionnel ; l’échocœur a objectivé une sténose de la valve pulmonaire et de l’APG.
Discussion :
Ces cas illustrent la diversité des CC syndromiques rares, liées à des anomalies embryologiques et génétiques. Le diagnostic néonatal systématique réduit la morbidité, particulièrement si les ressources du diagnostic génétique sont très limitées.
Conclusion :
Face aux faciès syndromiques inhabituels, explorer systématiquement une CC est impératif, comme le soulignent ces cas, pour une prise en charge multidisciplinaire optimale.